
Simon J. Gathercole Gospel of Thomas Cladogram and evolutionary tree
The Unsolved Murder of JonBenet Ramsey :: The Unsolved Murder of JonBenet Ramsey-BLOGS :: Redpill's Blog
Page 1 of 1

Simon J. Gathercole Gospel of Thomas Cladogram and evolutionary tree
![]() by redpill Tue Sep 04, 2018 7:58 pm
by redpill Tue Sep 04, 2018 7:58 pm
Tue Sep 04, 2018
time for sunday school with redpill
with redpill, every day is sunday school.
disclaimer i've not read Simon J. Gathercole book on Gospel of Thomas in its entirety, just some bits and pieces on google books
https://books.google.com/books?id=TWe7AwAAQBAJ&q=dating#v=snippet&q=dating&f=true

just bits and pieces as allowed by google books
but i've seen it discussed by christian debators and references by christian debators like josh mcdowell who cite his work as proof that the Gospel of Thomas is a late second century gnostic composition and of no relevance to historical jesus.
the conclusion of his work as cited by several footnotes is GThomas is late no relevance to historical jesus.
christian fundamentalists want to claim its late second century and therefore its contents are not for christian consumption, yet at the same time, will cite this as evidence that jesus existed, against certain atheists who claim jesus never existed.
since GThomas is just a collection of sayings, Simon J. Gathercole thesis is whether every single saying and must be dependent on Luke
and how he thinks gThomas dependence on Luke is the best explanation rather than Luke dependence on Thomas, both came from same source, or some other explanation.
I'm sure Simon J. Gathercole is a fine NT scholar, but certainly the field does attract fundamentalist christians who are committed to preserving an orthodoxy. one that states that GThomas is late, and at the same time, its evidence Jesus existed.
Christian fundamentalists don't quote Bart Ehrman for example, on the consensus Matthew and Luke used Mark. I don't think they are very intelligent persons, for the most part. Comparing Luke and Matthew with Mark side by side leads to the conclusion that yes they used Mark.
Simon J. Gathercole and others are trained in history.
What i think though with GThomas and other gospels could better benefit from is evolutionary analysis that is done in genetics and linguistics.
One set of topics is the cladogram
i'm arguing that linguists using an analysis that is analgous to the above, on the Gospel of Thomas, and the other gospels, mark matthew luke and John,
using science that is,
construct the most parsimonious trees to determine relationships among content among the gospels

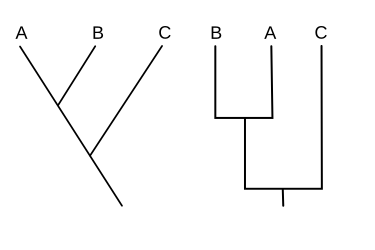
so Gathercole proposes that several sayings in Thomas are dependent on Luke,
I propose using science, specifically what is similar to what geneticists used to determine evolutionary relationships, to determine the most parsimonious tree.
Perhaps Luke is dependent on Thomas. Perhaps Luke and Thomas are dependent on a lost source.
use science wherever possible. It's possible Mark used Thomas, or that a cladogram of similar material in Q and Thomas can construct the original sayings Gospel.
this may require linguists and translators familiar with Aramaic, Jesus original language. and Greek, the language of the NT and Thomas.
in a perfect world of course would be to somehow find the original documents and date them directly but it appears such documents are forever lost.
In other words, the actual issue of dating GThomas, i argue, barring finding the original documents which would be something of a miracle,
would be to do Quantitative comparative linguistics
to use Gospel of Thomas and Gospel of Luke and Matthew and perform this sort of analysis
to determine how Thomas is related to Q and Matthew Mark Luke and John.
rather than an NT scholar using only historical but not scientifically verified methodology to speculate that Thomas used Matthew Mark Luke and John rather than the reverse, or use of multiple now lost sources
IMO many of the sayings when they are paralleled by the synoptic, seem much more primitive and simpler and therefore closer to the original.
time for sunday school with redpill
with redpill, every day is sunday school.
disclaimer i've not read Simon J. Gathercole book on Gospel of Thomas in its entirety, just some bits and pieces on google books
https://books.google.com/books?id=TWe7AwAAQBAJ&q=dating#v=snippet&q=dating&f=true
just bits and pieces as allowed by google books
but i've seen it discussed by christian debators and references by christian debators like josh mcdowell who cite his work as proof that the Gospel of Thomas is a late second century gnostic composition and of no relevance to historical jesus.
the conclusion of his work as cited by several footnotes is GThomas is late no relevance to historical jesus.
christian fundamentalists want to claim its late second century and therefore its contents are not for christian consumption, yet at the same time, will cite this as evidence that jesus existed, against certain atheists who claim jesus never existed.
since GThomas is just a collection of sayings, Simon J. Gathercole thesis is whether every single saying and must be dependent on Luke
and how he thinks gThomas dependence on Luke is the best explanation rather than Luke dependence on Thomas, both came from same source, or some other explanation.
I'm sure Simon J. Gathercole is a fine NT scholar, but certainly the field does attract fundamentalist christians who are committed to preserving an orthodoxy. one that states that GThomas is late, and at the same time, its evidence Jesus existed.
Christian fundamentalists don't quote Bart Ehrman for example, on the consensus Matthew and Luke used Mark. I don't think they are very intelligent persons, for the most part. Comparing Luke and Matthew with Mark side by side leads to the conclusion that yes they used Mark.
Simon J. Gathercole and others are trained in history.
What i think though with GThomas and other gospels could better benefit from is evolutionary analysis that is done in genetics and linguistics.
One set of topics is the cladogram
A cladogram (from Greek clados "branch" and gramma "character") is a diagram used in cladistics to show relations among organisms. A cladogram is not, however, an evolutionary tree because it does not show how ancestors are related to descendants, nor does it show how much they have changed; many evolutionary trees can be inferred from a single cladogram.[1][2][3][4][5] A cladogram uses lines that branch off in different directions ending at a clade, a group of organisms with a last common ancestor. There are many shapes of cladograms but they all have lines that branch off from other lines. The lines can be traced back to where they branch off. These branching off points represent a hypothetical ancestor (not an actual entity) which can be inferred to exhibit the traits shared among the terminal taxa above it.[4][6] This hypothetical ancestor might then provide clues about the order of evolution of various features, adaptation, and other evolutionary narratives about ancestors. Although traditionally such cladograms were generated largely on the basis of morphological characters, DNA and RNA sequencing data and computational phylogenetics are now very commonly used in the generation of cladograms, either on their own or in combination with morphology.
Molecular versus morphological data
The characteristics used to create a cladogram can be roughly categorized as either morphological (synapsid skull, warm blooded, notochord, unicellular, etc.) or molecular (DNA, RNA, or other genetic information).[7] Prior to the advent of DNA sequencing, cladistic analysis primarily used morphological data. Behavioral data (for animals) may also be used.[8]
As DNA sequencing has become cheaper and easier, molecular systematics has become a more and more popular way to infer phylogenetic hypotheses.[9] Using a parsimony criterion is only one of several methods to infer a phylogeny from molecular data. Approaches such as maximum likelihood, which incorporate explicit models of sequence evolution, are non-Hennigian ways to evaluate sequence data. Another powerful method of reconstructing phylogenies is the use of genomic retrotransposon markers, which are thought to be less prone to the problem of reversion that plagues sequence data. They are also generally assumed to have a low incidence of homoplasies because it was once thought that their integration into the genome was entirely random; this
Cladogram selection
There are several algorithms available to identify the "best" cladogram.[14] Most algorithms use a metric to measure how consistent a candidate cladogram is with the data. Most cladogram algorithms use the mathematical techniques of optimization and minimization.
In general, cladogram generation algorithms must be implemented as computer programs, although some algorithms can be performed manually when the data sets are modest (for example, just a few species and a couple of characteristics).
Some algorithms are useful only when the characteristic data are molecular (DNA, RNA); other algorithms are useful only when the characteristic data are morphological. Other algorithms can be used when the characteristic data includes both molecular and morphological data.
Algorithms for cladograms or other types of phylogenetic trees include least squares, neighbor-joining, parsimony, maximum likelihood, and Bayesian inference.
Biologists sometimes use the term parsimony for a specific kind of cladogram generation algorithm and sometimes as an umbrella term for all phylogenetic algorithms.[15]
Algorithms that perform optimization tasks (such as building cladograms) can be sensitive to the order in which the input data (the list of species and their characteristics) is presented. Inputting the data in various orders can cause the same algorithm to produce different "best" cladograms. In these situations, the user should input the data in various orders and compare the results.
Using different algorithms on a single data set can sometimes yield different "best" cladograms, because each algorithm may have a unique definition of what is "best".
Because of the astronomical number of possible cladograms, algorithms cannot guarantee that the solution is the overall best solution. A nonoptimal cladogram will be selected if the program settles on a local minimum rather than the desired global minimum.[16] To help solve this problem, many cladogram algorithms use a simulated annealing approach to increase the likelihood that the selected cladogram is the optimal one.[17]
The basal position is the direction of the base (or root) of a rooted phylogenetic tree or cladogram. A basal clade is the earliest clade (of a given taxonomic rank[a]) to branch within a larger clade.
Statistics
Incongruence length difference test (or partition homogeneity test)
The incongruence length difference test (ILD) is a measurement of how the combination of different datasets (e.g. morphological and molecular, plastid and nuclear genes) contributes to a longer tree. It is measured by first calculating the total tree length of each partition and summing them. Then replicates are made by making randomly assembled partitions consisting of the original partitions. The lengths are summed. A p value of 0.01 is obtained for 100 replicates if 99 replicates have longer combined tree lengths.
Measuring homoplasy
Further information: Convergent evolution
Some measures attempt to measure the amount of homoplasy in a dataset with reference to a tree,[18] though it is not necessarily clear precisely what property these measures aim to quantify[19]
Consistency index
The consistency index (CI) measures the consistency of a tree to a set of data – a measure of the minimum amount of homoplasy implied by the tree.[20] It is calculated by counting the minimum number of changes in a dataset and dividing it by the actual number of changes needed for the cladogram.[20] A consistency index can also be calculated for an individual character i, denoted ci.
Besides reflecting the amount of homoplasy, the metric also reflects the number of taxa in the dataset,[21] (to a lesser extent) the number of characters in a dataset,[22] the degree to which each character carries phylogenetic information,[23] and the fashion in which additive characters are coded, rendering it unfit for purpose.[24]
ci occupies a range from 1 to 1/[n.taxa/2] in binary characters with an even state distribution; its minimum value is larger when states are not evenly spread.[23][25]
Retention index
The retention index (RI) was proposed as an improvement of the CI "for certain applications"[26] This metric also purports to measure of the amount of homoplasy, but also measures how well synapomorphies explain the tree. It is calculated taking the (maximum number of changes on a tree minus the number of changes on the tree), and dividing by the (maximum number of changes on the tree minus the minimum number of changes in the dataset).
The rescaled consistency index (RC) is obtained by multiplying the CI by the RI; in effect this stretches the range of the CI such that its minimum theoretically attainable value is rescaled to 0, with its maximum remaining at 1.[25][26] The homoplasy index (HI) is simply 1 − CI.
Homoplasy Excess Ratio
This measures the amount of homoplasy observed on a tree relative to the maximum amount of homoplasy that could theoretically be present – 1 − (observed homoplasy excess) / (maximum homoplasy excess).[22] A value of 1 indicates no homoplasy; 0 represents as much homoplasy as there would be in a fully random dataset, and negative values indicate more homoplasy still (and tend only to occur in contrived examples).[22] The HER is presented as the best measure of homoplasy currently available.[25][27]
i'm arguing that linguists using an analysis that is analgous to the above, on the Gospel of Thomas, and the other gospels, mark matthew luke and John,
using science that is,
construct the most parsimonious trees to determine relationships among content among the gospels
so Gathercole proposes that several sayings in Thomas are dependent on Luke,
I propose using science, specifically what is similar to what geneticists used to determine evolutionary relationships, to determine the most parsimonious tree.
Perhaps Luke is dependent on Thomas. Perhaps Luke and Thomas are dependent on a lost source.
use science wherever possible. It's possible Mark used Thomas, or that a cladogram of similar material in Q and Thomas can construct the original sayings Gospel.
this may require linguists and translators familiar with Aramaic, Jesus original language. and Greek, the language of the NT and Thomas.
in a perfect world of course would be to somehow find the original documents and date them directly but it appears such documents are forever lost.
In other words, the actual issue of dating GThomas, i argue, barring finding the original documents which would be something of a miracle,
would be to do Quantitative comparative linguistics
to use Gospel of Thomas and Gospel of Luke and Matthew and perform this sort of analysis
During the 1950s, the Swadesh list emerged: a standardised set of lexical concepts found in most languages, as words or phrases, that allow two or more languages to be compared and contrasted empirically.
Probably the first published quantitative historical linguistics study was by Sapir in 1916,[1] while Kroeber and Chretien in 1937 [2] investigated nine Indo-European (IE) languages using 74 morphological and phonological features (extended in 1939 by the inclusion of Hittite). Ross [3] in 1950 carried out an investigation into the theoretical basis for such studies. Swadesh, using word lists, developed lexicostatistics and glottochronology in a series of papers [4] published in the early 1950s but these methods were widely criticised [5] though some of the criticisms were seen as unjustified by other scholars. Embleton published a book on "Statistics in Historical Linguistics" in 1986 which reviewed previous work and extended the glottochronological method. Dyen, Kruskal and Black carried out a study of the lexicostatistical method on a large IE database in 1992.[6]
During the 1990s, there was renewed interest in the topic, based on the application of methods of computational phylogenetics and cladistics. Such projects often involved collaboration by linguistic scholars, and colleagues with expertise in information science and/or biological anthropology. These projects often sought to arrive at an optimal phylogenetic tree (or network), to represent a hypothesis about the evolutionary ancestry and perhaps its language contacts. Pioneers in these methods included the founders of CPHL: computational phylogenetics in historical linguistics (CPHL project): Donald Ringe, Tandy Warnow, Luay Nakhleh and Steven N. Evans.
In the mid-1990s a group at Pennsylvania University computerised the comparative method and used a different IE database with 20 ancient languages.[7] In the biological field several software programs were then developed which could have application to historical linguistics. In particular a group at the University of Auckland developed a method that gave controversially old dates for IE languages.[8] A conference on "Time-depth in Historical Linguistics" was held in August 1999 at which many applications of quantitative methods were discussed.[9] Subsequently many papers have been published on studies of various language groups as well as comparisons of the methods.
Greater media attention was generated in 2003 after the publication by anthropologists Russell Gray and Quentin Atkinson of a short study on Indo-European languages in Nature. Gray and Atkinson attempted to quantify, in a probabilistic sense, the age and relatedness of modern Indo-European languages and, sometimes, the preceding proto-languages.
The proceedings of an influential 2004 conference, Phylogenetic Methods and the Prehistory of Languages were published in 2006, edited by Peter Forster and Colin Renfrew.
Studied language families
Computational phylogenetic analyses have been performed for:
Indo-European languages: Bouckaert (2012)[10]
Uralic languages: Honkola (2013)[11]
Turkic languages: Hruschka (2014)[12]
Dravidian languages: Kolipakam (2018)[13]
Austroasiatic languages: Sidwell (2015)[14]
Austronesian languages: Gray (2009)[15]
Pama-Nyungan languages: Bowern & Atkinson (2012),[16] Bouckaert (2018)[17]
Bantu languages: Currie (2013),[18] Grollemund (2015)[19]
Semitic languages: Kitchen (2009)[20]
Dené–Yeniseian languages: Sicoli & Holton (2014)[21]
Uto-Aztecan languages: Wheeler & Whiteley (2014)[22]
Mayan languages: Atkinson (2006)[23]
Arawakan languages: Walker & Ribeiro (2011)[24]
Tupi-Guarani languages: Michael (2015)[25]
Background
The standard method for assessing language relationships has been the comparative method. However this has a number of limitations. Not all linguistic material is suitable as input and there are issues of the linguistic levels on which the method operates. The reconstructed languages are idealized and different scholars can produce different results. Language family trees are often used in conjunction with the method and "borrowings" must be excluded from the data, which is difficult when borrowing is within a family. It is often claimed that the method is limited in the time depth over which it can operate. The method is difficult to apply and there is no independent test.[26] Thus alternative methods have been sought that have a formalised method, quantify the relationships and can be tested.
A goal of comparative historical linguistics is to identify instances of genetic relatedness amongst languages.[27] The steps in quantitative analysis are (i) to devise a procedure based on theoretical grounds, on a particular model or on past experience, etc. (ii) to verify the procedure by applying it to some data where there exists a large body of linguistic opinion for comparison (this may lead to a revision of the procedure of stage (i) or at the extreme of its total abandonment) (iii) to apply the procedure to data where linguistic opinions have not yet been produced, have not yet been firmly established or perhaps are even in conflict.[28]
Applying phylogenetic methods to languages is a multi-stage process: (a) the encoding stage - getting from real languages to some expression of the relationships between them in the form of numerical or state data, so that those data can then be used as input to phylogenetic methods (b) the representation stage - applying phylogenetic methods to extract from those numerical and/or state data a signal that is converted into some useful form of representation, usually two dimensional graphical ones such as trees or networks, which synthesise and "collapse" what are often highly complex multi dimensional relationships in the signal (c) the interpretation stage - assessing those tree and network representations to extract from them what they actually mean for real languages and their relationships through time.[29]
Types of trees and networks
An output of a quantitative historical linguistic analysis is normally a tree or a network diagram. This allows summary visualisation of the output data but is not the complete result. A tree is a connected acyclic graph, consisting of a set of vertices (also known as "nodes") and a set of edges ("branches") each of which connects a pair of vertices.[30] An internal node represents a linguistic ancestor in a phylogenic tree or network. Each language is represented by a path, the paths showing the different states as it evolves. There is only one path between every pair of vertices. Unrooted trees plot the relationship between the input data without assumptions regarding their descent. A rooted tree explicitly identifies a common ancestor, often by specifying a direction of evolution or by including an "outgroup" that is known to be only distantly related to the set of languages being classified. Most trees are binary, that is a parent has two children. A tree can always be produced even though it is not always appropriate. A different sort of tree is that only based on language similarities / differences. In this case the internal nodes of the graph do not represent ancestors but are introduced to represent the conflict between the different splits ("bipartitions") in the data analysis. The "phenetic distance" is the sum of the weights (often represented as lengths) along the path between languages. Sometimes an additional assumption is made that these internal nodes do represent ancestors.
When languages converge, usually with word adoption ("borrowing"), a network model is more appropriate. There will be additional edges to reflect the dual parentage of a language. These edges will be bidirectional if both languages borrow from one another. A tree is thus a simple network, however there are many other types of network. A phylogentic network is one where the taxa are represented by nodes and their evolutionary relationships are represented by branches.[31] Another type is that based on splits, and is a combinatorial generalisation of the split tree. A given set of splits can have more than one representation thus internal nodes may not be ancestors and are only an "implicit" representation of evolutionary history as distinct from the "explicit" representation of phylogenetic networks. In a splits network the phrenetic distance is that of the shortest path between two languages. A further type is the reticular network which shows incompatibilities (due to for example to contact) as reticulations and its internal nodes do represent ancestors. A network may also be constructed by adding contact edges to a tree. The last main type is the consensus network formed from trees. These trees may be as a result of bootstrap analysis or samples from a posterior distribution.
Language change
Change happens continually to languages, but not usually at a constant rate,[32] with its cumulative effect producing splits into dialects, languages and language families. It is generally thought that morphology changes slowest and phonology the quickest. As change happens, less and less evidence of the original language remains. Finally there could be loss of any evidence of relatedness. Changes of one type may not affect other types, for example sound changes do not affect cognancy. Unlike biology, it cannot be assumed that languages all have a common origin and establishing relatedness is necessary. In modelling it is often assumed for simplicity that the characters change independently but this may not be the case. Besides borrowing, there can also be semantic shifts and polymorphism.
Analysis input
Data
Analysis can be carried out on the "characters" of languages or on the "distances" of the languages. In the former case the input to a language classification generally takes the form of a data matrix where the rows correspond to the various languages being analysed and the columns correspond to different features or characters by which each language may be described. These features are of two types cognates or typological data. Characters can take one or more forms (homoplasy) and can be lexical, morphological or phonological. Cognates are morphemes (lexical or grammatical) or larger constructions. Typological characters can come from any part of the grammar or lexicon. If there are gaps in the data these have to be coded.
In addition to the original database of (unscreened) data, in many studies subsets are formed for particular purposes (screened data).
In lexicostatistics the features are the meanings of words, or rather semantic slots. Thus the matrix entries are a series of glosses. As originally devised by Swadesh the single most common word for a slot was to be chosen, which can be difficult and subjective because of semantic shift. Later methods may allow more than one meaning to be incorporated.
Constraints
Some methods allow constraints to be placed on language contact geography (isolation by distance) and on sub-group split times.
Databases
Swadesh originally published a 200 word list but later refined it into a 100 word one.[33] A commonly used IE database is that by Dyen, Kruskal and Black which contains data for 95 languages, though the original is known to contain a few errors. Besides the raw data it also contains cognacy judgements. This is available online.[34] The database of Ringe, Warnow and Taylor has information on 24 IE languages, with 22 phonological characters, 15 morphological characters and 333 lexical characters. Gray and Atkinson used a database of 87 languages with 2449 lexical items, based on the Dyen set with the addition of three ancient languages. They incorporated the cognacy judgements of a number of scholars. Other databases have been drawn up for African, Australian and Andean language families, amongst others.
Coding of the data may be in binary form or in multistate form. The former is often used but does result in a bias. It has been claimed that there is a constant scale factor between the two coding methods, and that allowance can be made for this. However, another study suggests that the topology may change [35]
Word lists
The word slots are chosen to be as culture- and borrowing- free as possible. The original Swadesh lists are most commonly used but many others have been devised for particular purposes. Often these are shorter than Swadesh's preferred 100 item list. Kessler has written a book on "The Significance of Word Lists [36] while McMahon and McMahon carried out studies on the effects of reconstructability and retentiveness.[37] The effect of increasing the number of slots has been studied and a law of diminishing returns found, with about 80 being found satisfactory.[38] However some studies have used less than half this number.
Generally each cognate set is represented as a different character but differences between words can also be measured as a distance measurement by sound changes. Distances may also be measured letter by letter.
Morphological features
Traditionally these have been seen as more important than lexical ones and so some studies have put additional weighting on this type of character. Such features were included in the Ringe, Warnow and Taylor IE database for example. However other studies have omitted them.
Typological features
Examples of these features include glottalised constants, tone systems, accusative alignment in nouns, dual number, case number correspondence, object-verb order, and first person singular pronouns. These will be listed in the WALS database, though this is only sparsely populated for many languages yet.[39]
Probabilistic models
Some analysis methods incorporate a statistical model of language evolution and use the properties of the model to estimate the evolution history. Statistical models are also used for simulation of data for testing purposes. A stochastic process can be used to describe how a set of characters evolves within a language. The probability with which a character will change can depend on the branch but not all charters evolve together, nor is the rate identical on all branches. It is often assumed that each character evolves independently but this is not always the case. Within a model borrowing and parallel development (homoplasy) may also be modelled, as well as polymorphisms.
Effects of chance
Chance resemblances produce a level of noise against which the required signal of relatedness has to be found. A study was carried out by Ringe [40] into the effects of chance on the mass comparison method. This showed that chance resemblances were critical to the technique and that Greenberg's conclusions could not be justified, though the mathematical procedure used by Rimge was later criticised.
With small databases sampling errors can be important.
In some cases with a large database and exhaustive search of all possible trees or networks is not feasible because of running time limitations. Thus there is a chance that the optimum solution is not found by heuristic solution-space search methods.
Detection of borrowing
Loanwords can severely affect the topology of a tree so efforts are made to exclude borrowings. However, undetected ones sometimes still exist. McMahon and McMahon [41] showed that around 5% borrowing can affect the topology while 10% has significant effects. In networks borrowing produces reticulations. Minett and Wang [42] examined ways of detecting borrowing automatically.
Split dating
Dating of language splits can be determined if it is known how the characters evolve along each branch of a tree. The simplest assumption is that all characters evolve at a single constant rate with time and that this is independent of the tree branch. This was the assumption made in glottochronology. However, studies soon showed that there was variation between languages, some probably due to the presence of unrecognised borrowing.[43] A better approach is to allow rate variation, and the gamma distribution is usually used because of its mathematical convenience. Studies have also been carried out that show that the character replacement rate depends on the frequency of use.[44] Widespread borrowing can bias divergence time estimates by making languages seem more similar and hence younger. However, this also makes the ancestor's branch length longer so that the root is unaffected.[45]
This aspect is the most controversial part of quantitative comparative linguistics.
Types of analysis
There is a need to understand how a language classification method works in order to determine its assumptions and limitations. It may only be valid under certain conditions or be suitable for small databases. The methods differ in their data requirements, their complexity and running time. The methods also differ in their optimisation criteria.
Character based models
Maximum parsimony and maximum compatibility
These two methods are similar but the maximum parsimony method's objective is to find the tree (or network) in which the minimum number of evolutionary changes occurs. In some implementations the characters can be given weights and then the objective is to minimise the total weighted sum of the changes. The analysis produces unrooted trees unless an outgroup is used or directed characters. Heuristics are used to find the best tree but optimisation is not guaranteed. The method is often implemented using the programs PAUP or TNT.
Maximum compatibility also uses characters, with the objective of finding the tree on which the maximum number of characters evolve without homoplasy. Again the characters can be weighted and when this occurs the objective is to maximise the sum of the weights of compatible characters. It also produces unrooted trees unless additional information is incorporated. There are no readily available heuristics available that are accurate with large databases. This method has only been used by Ringe's group.[46]
In these two methods there are often several trees found with the same score so the usual practice is to find a consensus tree via an algorithm. A majority consensus has bipartitions in more than half of the input trees while a greedy consensus adds bipartitions to the majority tree. The strict consensus tree is the least resolved and contains those splits that are in every tree.
Bootstrapping (a statistical resampling strategy) is used to provide branch support values. The technique randomly picks characters from the input data matrix and then the same analysis is used. The support value is the fraction of the runs with that bipartition in the observed tree. However, bootstrapping is very time consuming.
Maximum likelihood and Bayesian analysis
Both of these methods use explicit evolution models. The maximum likelihood method optimises the probability of producing the observed data, while Bayesian analysis estimates the probability of each tree and so produces a probability distribution. A random walk is made through the "model-tree space". Both take an indeterminate time to run, and stopping may be arbitrary so a decision is a problem. However, both produce support information for each branch.
The assumptions of these methods are overt and are verifiable. The complexity of the model can be increased if required. The model parameters are estimated directly from the input data so assumptions about evolutionary rate are avoided.
Perfect Phylogenetic Networks
This method produces an explicit phylogenic network having an underlying tree with additional contact edges. Characters can be borrowed but evolve without homoplasy. To produce such networks, a graph-theoretic algorithm [47] has been used.
Gray and Atkinson's method
The input lexical data is coded in binary form, with one character for each state of the original multi-state character. The method allows homoplasy and constraints on split times. A likelihood-based analysis method is used, with evolution expressed as a rate matrix. Cognate gain and loss is modelled with a gamma distribution to allow rate variation and with rate smoothing. Because of the vast number of possible trees with many languages, Bayesian inference is used to search for the optimal tree. A Markov Chain Monte Carlo algorithm [48] generates a sample of trees as an approximation to the posterior probability distribution. A summary of this distribution can be provided as a greedy consensus tree or network with support values. The method also provides date estimates.
The method is accurate when the original characters are binary, and evolve identically and independently of each other under a rates-across-sites model with gamma distributed rates; the dates are accurate when the rate of change is constant. Understanding the performance of the method when the original characters are multi-state is more complicated, since the binary encoding produces characters that are not independent, while the method assumes independence.
Nicholls and Gray's method
This method [49] is an outgrowth of Gray and Atkinson's. Rather than having two parameters for a character, this method uses three. The birth rate, death rate of a cognate are specified and its borrowing rate. The birth rate is a Poisson random variable with a single birth of a cognate class but separate deaths of branches are allowed (Dollo parsimony). The method does not allow homoplasy but allows polymorphism and constraints. Its major problem is that it cannot handle missing data (this issue has since been resolved by Ryder and Nicholls.[50] Statistical techniques are used to fit the model to the data. Prior information may be incorporated and an MCMC research is made of possible reconstructions. The method has been applied to Gray and Nichol's database and seems to give similar results.
Distance based models
These use a triangular matrix of pairwise language comparisons. The input character matrix is used to compute the distance matrix either using the Hamming distance or the Levenshtein distance. The former measures the proportion of matching characters while the latter allows costs of the various possible transforms to be included. These methods are fast compared with wholly character based ones. However, these methods do result in information loss.
UPGMA
The "Unweighted Pairwise Group Method with Arithmetic-mean" (UPGMA) is a clustering technique which operates by repeatedly joining the two languages that have the smallest distance between them. It operates accurately with clock-like evolution but otherwise it can be in error. This is the method used in Swadesh's original lexicostatistics.
Split Decomposition
This is a technique for dividing data into natural groups.[51] The data could be characters but is more usually distance measures. The character counts or distances are used to generate the splits and to compute weights (branch lengths) for the splits. The weighted splits are then represented in a tree or network based on minimising the number of changes between each pair of taxa. There are fast algorithms for generating the collection of splits. The weights are determined from the taxon to taxon distances. Split decomposition is effective when the number of taxa is small or when the signal is not too complicated.
Neighbor joining
This method operates on distance data, computes a transformation of the input matrix and then computes the minimum distance of the pairs of languages.[52] It operates correctly even if the languages do not evolve with a lexical clock. A weighted version of the method may also be used. The method produces an output tree. It is claimed to be the closest method to manual techniques for tree construction.
Neighbor-net
It uses a similar algorithm to neighbor joining.[53] Unlike Split Decomposition it does not fuse nodes immediately but waits until a node has been paired a second time. The tree nodes are then replaced by two and the distance matrix reduced. It can handle large and complicated data sets. However, the output is a phenogram rather than a phylogram. This is the most popular network method.
Network
This was an early network method that has been used for some language analysis. It was originally developed for genetic sequences with more than one possible origin.[54] Network collapses the alternative trees into a single network. Where there are multiple histories a reticulation (a box shape) is drawn. It generates a list of characters incompatible with a tree.
ASP
This uses a declarative knowledge representation formalism and the methods of Answer Set Programming.[55] One such solver is CMODELS which can be used for small problems but larger ones require heuristics. Preprocessing is used to determine the informative characters. CMODELS transforms them into a propositional theory that uses a SAT solver to compute the models of this theory.
Fitch/Kitch
Fitch and Kitch are maximum likelihood based programs in PHYLIP that allow a tree to be rearranged after each addition, unlike NJ. Kitch differs from Fitch in assuming a constant rate of change throughout the tree while Fitch allows for different rates down each branch.[56]
Separation level method
Holm introduced a method in 2000 to deal with some known problems of lexicostatistical analysis. These are the "symplesiomorphy trap", where shared archaisms are difficult to distinguish from shared innovations, and the "proportionality "trap" when later changes can obscure early ones. Later he introduced a refined method, called SLD, to take account of the variable word distribution across languages.[57] The method does not assume aconstant rate of change.
to determine how Thomas is related to Q and Matthew Mark Luke and John.
rather than an NT scholar using only historical but not scientifically verified methodology to speculate that Thomas used Matthew Mark Luke and John rather than the reverse, or use of multiple now lost sources
IMO many of the sayings when they are paralleled by the synoptic, seem much more primitive and simpler and therefore closer to the original.
_________________
If you only knew the POWER of the Daubert side

redpill- Posts : 6216
Join date : 2012-12-08
» Simon J. Gathercole Gospel of Thomas and Forensic Linguistics
» What christians say about Gospel of Thomas saying 113 and how it applies to the New Testament
» there was an older version of Gospel of Thomas which was Q & Matthew and Luke used it
» John Ankerberg Gospel of Thomas vs Jesus in Revelation
» gospels of matthew, luke, mark and gospel of thomas christian special pleading
» What christians say about Gospel of Thomas saying 113 and how it applies to the New Testament
» there was an older version of Gospel of Thomas which was Q & Matthew and Luke used it
» John Ankerberg Gospel of Thomas vs Jesus in Revelation
» gospels of matthew, luke, mark and gospel of thomas christian special pleading
The Unsolved Murder of JonBenet Ramsey :: The Unsolved Murder of JonBenet Ramsey-BLOGS :: Redpill's Blog
Page 1 of 1
Permissions in this forum:
You cannot reply to topics in this forum|
|
|